Research
My research focuses on developing and applying computational methods to understand the electronic and optical properties of emerging quantum materials. I work on method development, band-structure modeling, and exciton physics in low-dimensional systems.
Semi-Empirical Pseudopotential Method (SEPM)
A major part of my work is the development and application of the Semi-Empirical Pseudopotential Method (SEPM). This approach provides an efficient and accurate way to compute electronic structures for large and complex systems. In our implementation, the in-plane coordinates are treated with plane waves, while the out-of-plane direction is expanded using B-spline basis functions. The method includes local and non-local potential terms and is parameterized to reproduce key results from first-principles Density Functional Theory (DFT) calculations and, where available, experimental data.
This work is carried out in collaboration with Prof. Chung-Yuan Ren (NKNU).
Applications to Graphene and Graphene Nanoribbons
We have applied SEPM to graphene and armchair graphene nanoribbons (aGNRs), capturing the Dirac-cone physics of graphene and the width-dependent band gaps of nanoribbons. These results are useful for understanding nanoelectronic behavior in carbon-based materials. Related work is reported in Semi-Empirical Pseudopotential Method for Graphene and Graphene Nanoribbons.
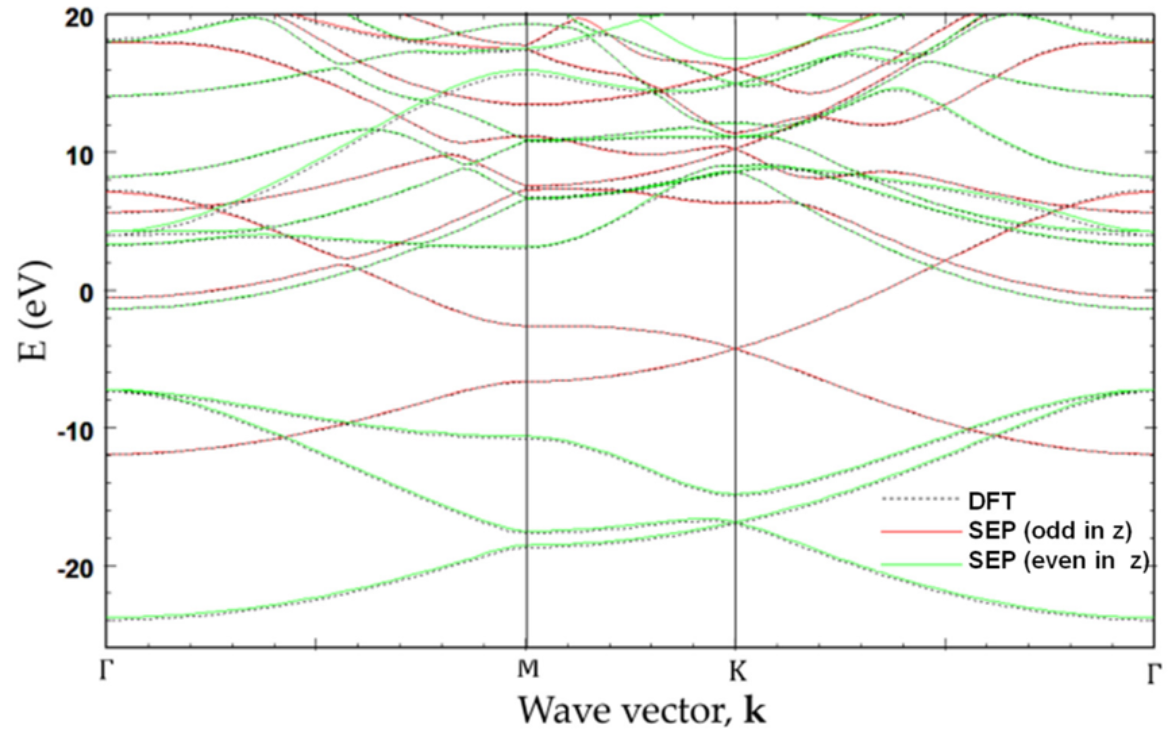
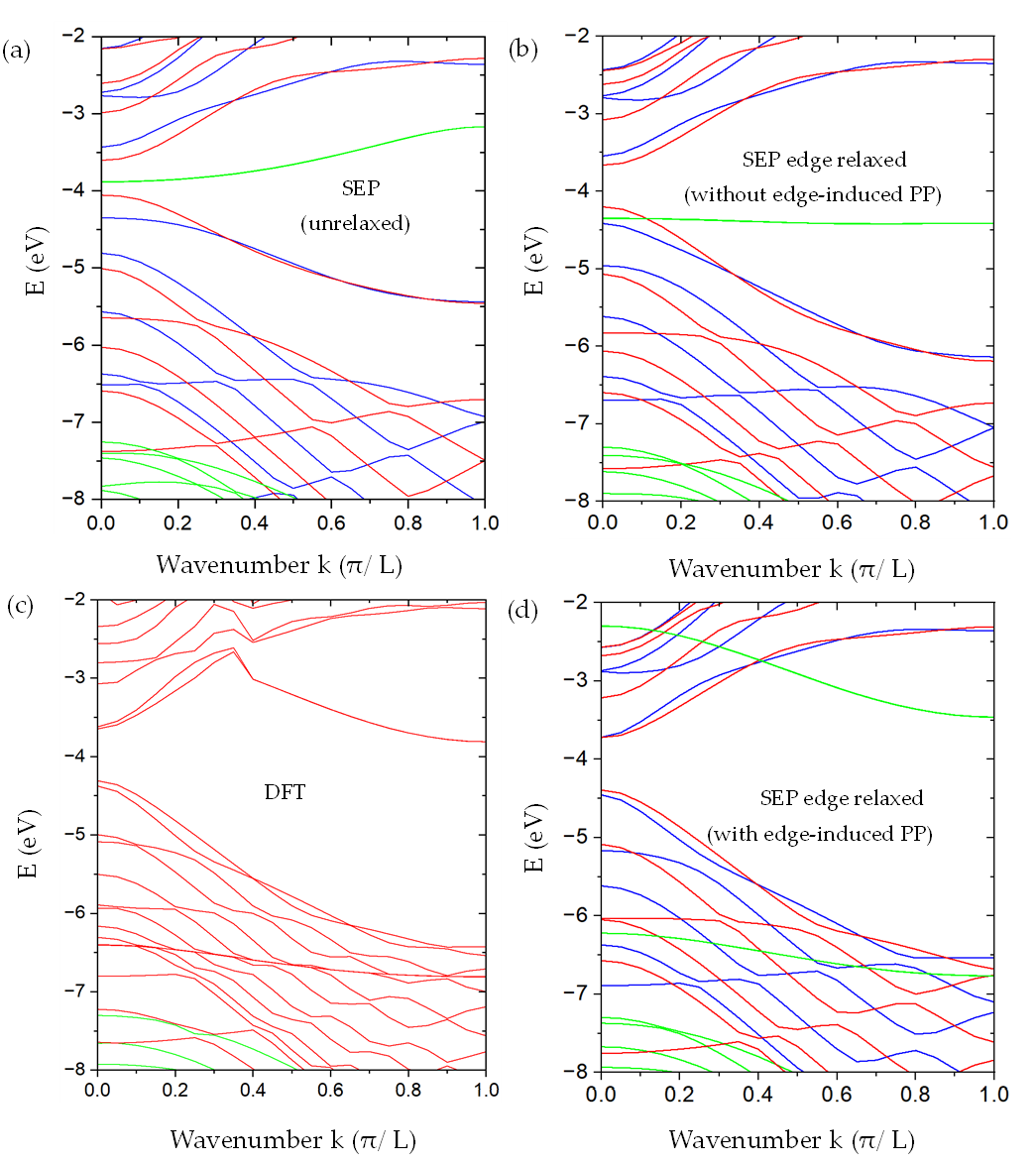
Applications to Transition-Metal Dichalcogenides (TMDCs)
The SEPM framework has also been extended to monolayer transition-metal dichalcogenides (TMDCs). This provides a fast and reliable way to calculate band structures and to study the electronic ingredients that influence optical response and exciton formation. A preprint is available here: Semiempirical Pseudopotential Method for Transitional-Metal Dichalcogenides.
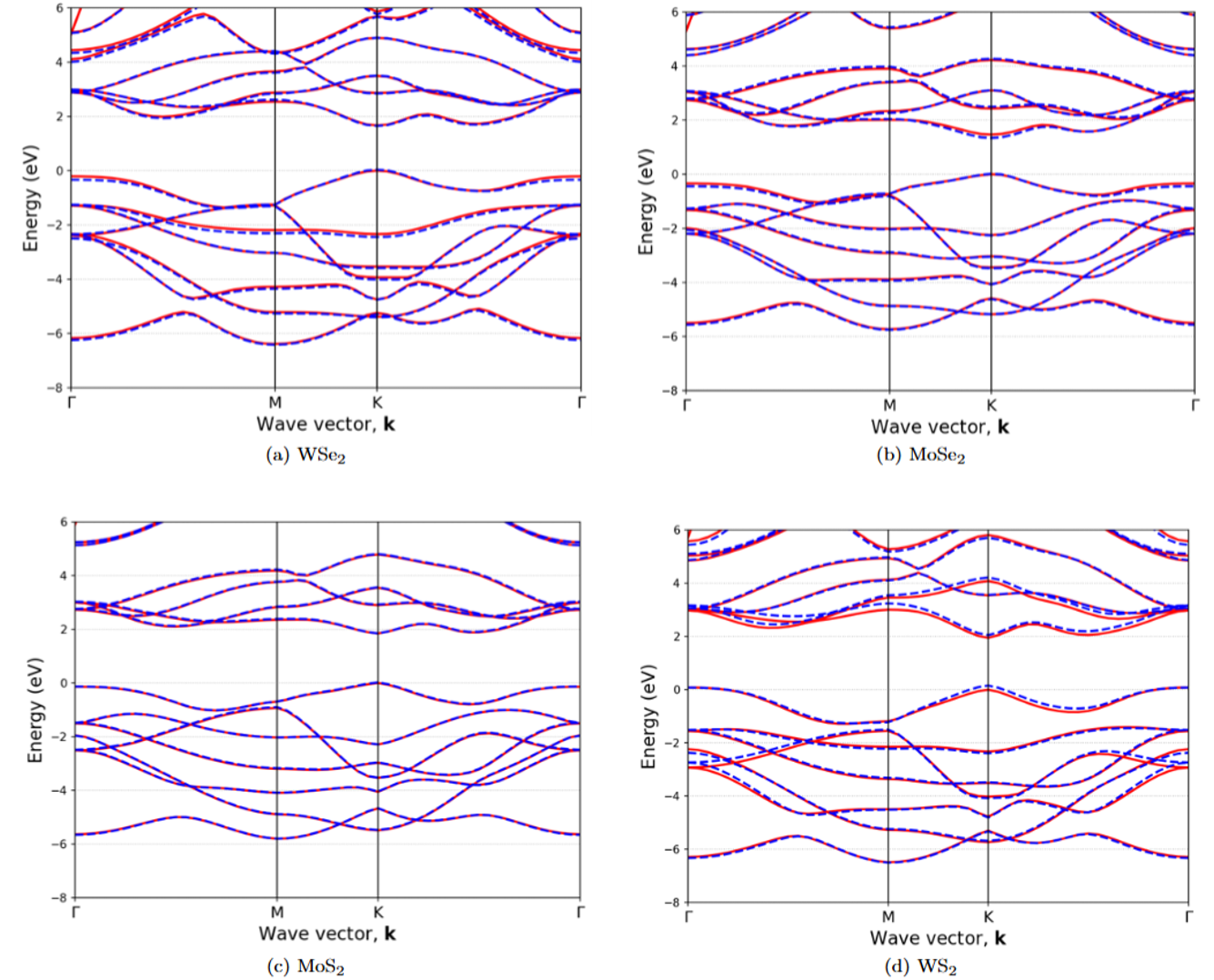
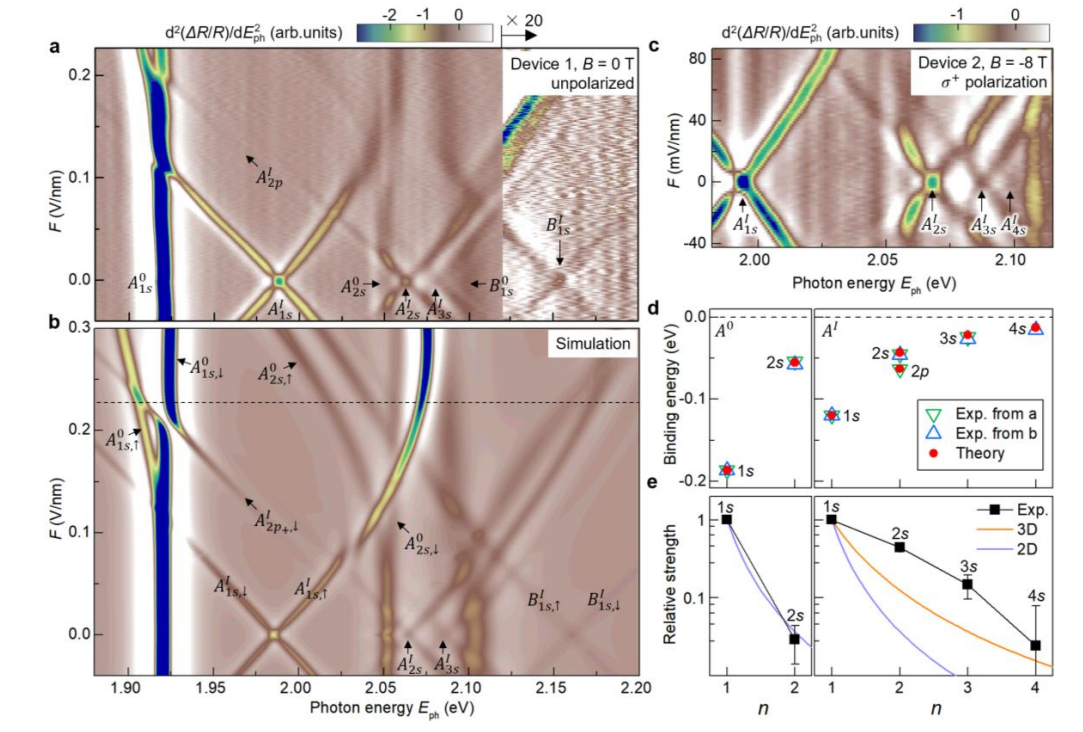
Exciton Physics in TMDCs
Monolayer and few-layer TMDCs exhibit strong light-matter interaction dominated by excitons, which are bound electron-hole pairs. My research in this area focuses on exciton binding energies, interlayer and intralayer excitons, electric-field control, and hybridization effects. These systems are important for next-generation optoelectronic and valleytronic devices.
- Interlayer and intralayer excitons in monolayer and bilayer TMDCs
- Electric-field-driven brightening and charge redistribution
- 2p interlayer exciton states and hybridization effects
Transport and Charge Transfer in F4TCNQ-Graphene Systems
A current direction involves studying charge transfer and transport properties of F4TCNQ molecules adsorbed on graphene. Using large-scale graphene supercells (e.g., a 16 × 16 graphene supercell), we investigate the modification of graphene's electronic structure, charge redistribution, and transport characteristics induced by molecular adsorption.
The calculations combine density functional theory with quantum transport methods to analyze transmission spectra, conductance behavior, and the influence of molecular dopants on graphene-based electronic devices. This work is carried out in collaboration with Prof. Ming-Hao Liu.
- DFT-based electronic structure calculations of molecule–graphene interfaces
- Charge density difference and electron transfer analysis
- NEGF-based transmission and conductance calculations
- Design principles for molecularly functionalized graphene devices
Future Directions
Future research will focus on developing efficient computational frameworks that bridge electronic structure calculations, many-body physics, and quantum transport in emerging low-dimensional materials. By combining semi-empirical pseudopotential methods (SEPM), first-principles calculations, and quantum transport approaches, I aim to study complex quantum systems beyond the limitations of conventional computational methods.
One important direction is extending SEPM toward large-scale two-dimensional materials and van der Waals heterostructures. Accurate modeling of interlayer coupling, moiré potentials, and electric-field effects will enable the investigation of novel electronic phases and excitonic phenomena in twisted bilayer systems.
Another direction involves connecting material modeling with device applications through quantum transport calculations. By combining density functional theory with nonequilibrium Green's function (NEGF) methods, future work will explore charge transfer, molecular doping, and transport properties of functionalized 2D materials, including graphene-based molecular interfaces.
Ultimately, the goal is to develop computational tools that can efficiently predict electronic, optical, and transport properties of quantum materials and guide the design of next-generation nanoelectronic and optoelectronic devices.